
Prof. Jun Ma (Sun Yat-sen Universiteit) presenteerde de resultaten van een multicentrische, open-label, gerandomiseerde fase III-studie die radiotherapie vergeleek met gelijktijdige chemoradiotherapie bij het nasofaryngeaal carcinoom (NPC) met gemiddeld risico.
Radiotherapie vs. chemoradiotherapie bij nasofaryngeaal carcinoom
Gelijktijdige chemoradiotherapie (CCRT) met of zonder inductie of adjuvante chemotherapie is momenteel de standaardbehandeling voor stadium II NPC, gebaseerd op een gerandomiseerde studie waarbij tweedimensionale conventionele radiotherapie werd gebruikt. Maar in tijden van Intensity-Modulated Radiation Therapy (IMRT) beschikken we nog niet over voldoende bewijs over de rol van chemotherapie bij deze populatie. Bovendien verhoogde een op cisplatinum gebaseerde CCRT ernstige acute nevenwerkingen, latere toxiciteit en een risico op behandelingsgerelateerd overlijden.
Een studie bij patiënten met stadium II- of T3N0-NPC toonde daarnaast aan dat de toevoeging van gelijktijdige chemotherapie aan IMRT geen significant overlevingsvoordeel opleverde (1).
Deze multicentrische, gerandomiseerde klinische fase III-studie moet bewijzen of het weglaten van de gelijktijdige chemotherapie wel veilig is voor patiënten met laag-risico NPC behandeld met IMRT. Laag-risico NPC werd gedefinieerd als stadium II- of T3N0M0-ziekte zonder de volgende gekende ongunstige kenmerken: maximale diameter van een cervicale lymfeklier > 3 cm, level IV of Vb lymfeklieren, extranodale extensie of een Epstein Barr-virus DNA-detectie > 4.000 kopieën/mL.
De studie randomiseerde 341 laag-risico NPC-patiënten naar enkel IMRT (n = 172) of naar IMRT in combinatie met gelijktijdig cisplatinum 100mg/m2 IV op dag 1, 22 en 43 (n = 169). Het primaire eindpunt van de studie is failure-free survival (FFS) gedefinieerd als de tijd tot recidief (zowel locoregionaal als op afstand) of tot overlijden. Secundaire eindpunten zijn onder andere globale overleving (OS), het veiligheidsprofiel en levenskwaliteit (HRQoL). De mediane follow-up bedroeg 46 maanden. In de CCRT-groep kregen 60,4% van de patiënten drie cycli van gelijktijdige chemotherapie. 97,3% kreeg ten minste twee cycli.
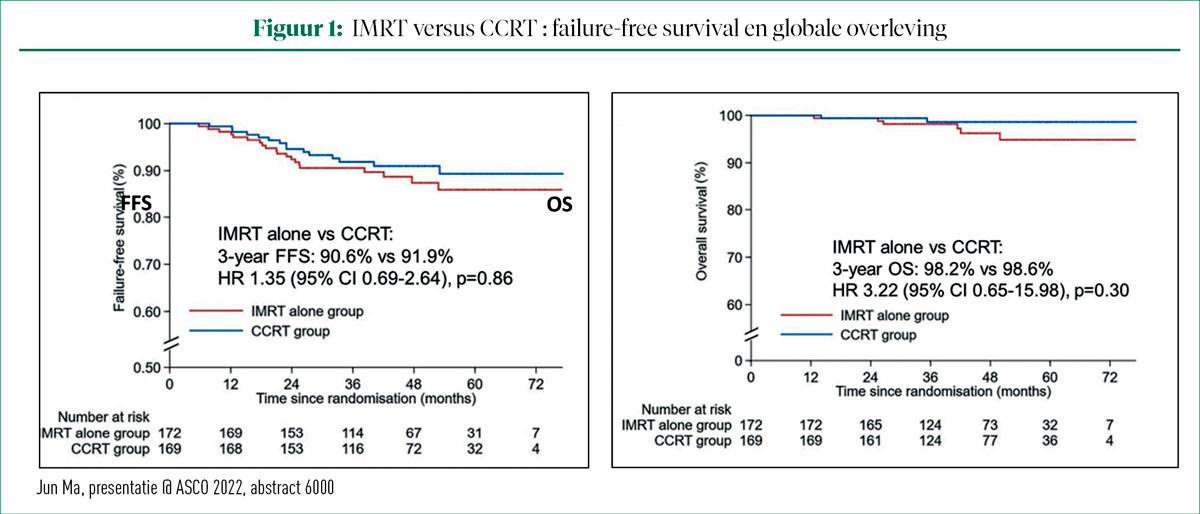
Analyse van het primaire eindpunt FFS laat zien dat de groep met alleen IMRT een nagenoeg identieke driejarige FFS toont in vergelijking met de CCRT-groep. De driejarige FFS is respectievelijk 98,6% en 91,9%, wat overeenkwam met een non-inferioriteitsmarge van minder dan 10%. De OS na drie jaar was 98,2% en 97,6% voor respectievelijk de IMRT-groep en de CCRT-groep (zie figuur 1).
Gedurende de gehele behandelingskuur was er een significant lagere incidentie van gerapporteerde nevenwerkingen van graad 3-4 in de groep met alleen IMRT (17%) vergeleken met de CCRT-groep (46,2%). Ook levenskwaliteit, gemeten met de EORTC QLQ-C30 scoort significant beter voor een behandeling met IMRT alleen met duidelijke verschillen voor vermoeidheid, misselijkheid en braken, pijn, slapeloosheid, verminderde eetlust en constipatie.
Prof. Jun Ma concludeerde dan ook dat enkel IMRT bij patiënten met een stadium II en T3N0 nasofaryngeaal carcinoom een volwaardige behandelingsoptie is. Deze behandeling zorgt voor een niet-inferieure ziektecontrole, verminderde toxiciteit en een verbeterde kwaliteit van leven (2).
Docetaxel als radiosensitizer
Dr. Vanita Noronha (Tata Memorial Hospital, Mumbai, India) presenteerde resultaten van een gerandomiseerde fase III-studie over het gebruik van docetaxel als radiosensitizer bij patiënten met hoofd- en halskanker die niet geschikt zijn voor op cisplatinum (CIS) gebaseerde chemoradiotherapie. Chemoradiotherapie op basis van CIS is de standaardbehandeling voor patiënten met lokaal gevorderd plaveiselcelcarcinoom van het hoofd- en halsgebied. Een aantal patiënten is niet geschikt om CIS te krijgen. En er zijn echt zeer beperkte gegevens over een alternatieve behandeling bij deze patiënten. Fase I- en II-studies met docetaxel (DOX) waren veelbelovend en bijna alle patiënten die niet geschikt zijn voor CIS kunnen docetaxel krijgen. Gegevens uit fase III-studies zijn beschikbaar voor cetuximab, carboplatinum en 5FU, carboplatinum in monotherapie, en de combinatie van paclitaxel en carboplatinum. Al deze fase III-gegevens zijn echter afkomstig van patiëntencohortes die in origine wel in aanmerking kwamen voor CIS. Vandaar de nood om de rol van DOX te onderzoeken bij patiënten die niet in aanmerking komen voor CIS.
Deze fase III-studie includeerde volwassen patiënten met een ECOG-performantiestatus 0 tot 2, met lokaal gevorderd plaveiselcelcarcinoom van het hoofd- en halsgebied, die gepland waren voor chemoradiotherapie in de adjuvante of in de definitieve setting. Patiënten moesten CIS-ongeschikt zijn. Het niet in aanmerking komen voor een CIS-behandeling werd gedefinieerd als de aanwezigheid van één of meerdere van de volgende criteria: een ECOG-performantiestatus gelijk aan twee; een orgaandisfunctie hoger dan graad 2 volgens CTCAE versie 4 (Common Terminology Criteria for Adverse Events) zoals bijvoorbeeld gehoorverlies, tinnitus of neurologische aandoeningen; overgevoeligheid voor CIS; een berekende creatinineklaring van minder dan 50 ml per minuut, een borderline functioneren van een orgaan, of de aanwezigheid van comorbiditeiten die het gebruik van CIS uitsloten; verlies van meer dan 10% van het lichaamsgewicht in de voorafgaande zes maanden; een ondervoede status gedefinieerd door een BMI < 16 kg/m2 en het gelijktijdig gebruik van nefrotoxische geneesmiddelen nodig voor de behandeling van een samenlopende medische aandoening.
Patiënten werden vervolgens een op een gerandomiseerd naar alleen radiotherapie (RT) of naar de combinatie van DOX met RT. DOX werd eenmaal per week gedoseerd aan 15 mg/m2 gedurende de volledige radiotherapie. Eindpunten van de studie waren PFS na twee jaar, OS na twee jaar, QoL en de bijwerkingen.
De rekrutering van deze studie verliep traag omdat de studie werd uitgevoerd midden in de covidpandemie. Uiteindelijk werden 777 patiënten beoordeeld op geschiktheid en 356 gerandomiseerd, 176 naar de arm met alleen RT en 180 naar de arm met DOX + RT. De meerderheid van de patiënten had stadium 4A. De HPV-status werd gecontroleerd bij ongeveer de helft van de patiënten met een orofaryngeale primaire tumor, waarbij het HPV-positiviteitspercentage 4% bedraagt. Van de patiënten kreeg 60% chemoradiotherapie in de definitieve behandelingssetting en ongeveer 40% van de patiënten kreeg het in de adjuvante setting. Een kwart van de patiënten kwam niet in aanmerking voor een behandeling met CIS vanwege een lage creatinineklaring. Ongeveer 40% van de patiënten had perceptief gehoorverlies en ongeveer 40% van de patiënten had een ECOG-performantiestatus gelijk aan twee. De meeste van de patiënten vertoonden trouwens verschillende redenen waarom ze niet in aanmerking kwamen voor CIS. Meer dan 90% van de patiënten kreeg 100% van de RT-dosis met onderbrekingen van de bestraling in de DOX-arm bij 11% van de patiënten.
Het mediane aantal afgeleverde cycli van DOX was zes. Meer dan 80% van de patiënten kreeg meer dan vijf cycli. Wat betreft bijwerkingen, verhoogde de toevoeging van DOX aan RT significant het aantal patiënten met mucositis, odynofagie en dysfagie. De andere toxiciteiten, zoals dermatitis, misselijkheid, braken en febriele neutropenie werden niet verhoogd door het toevoegen van DOX.
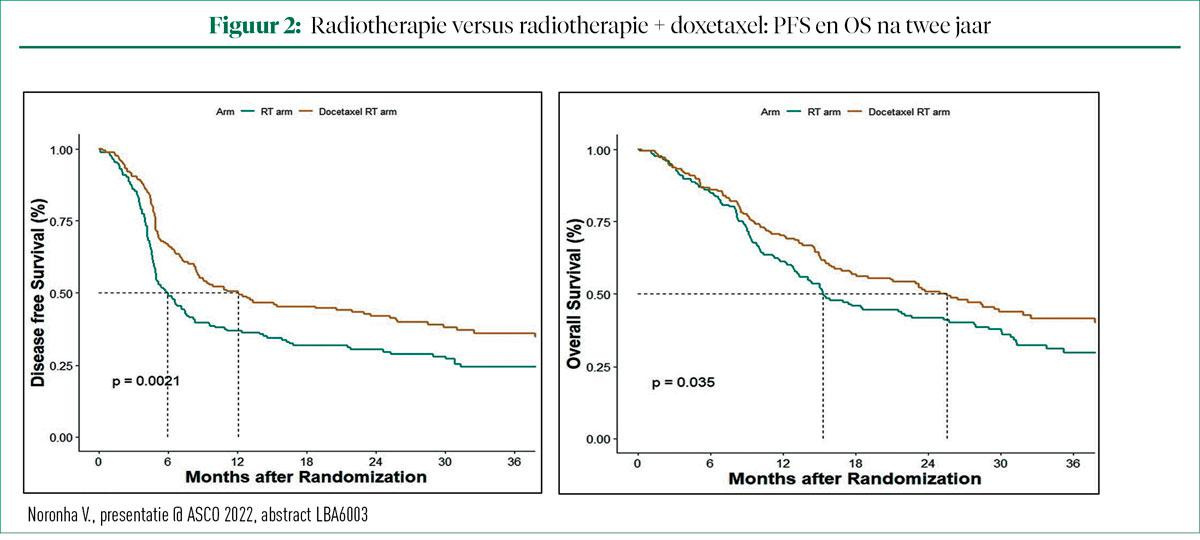
Bij patiënten die alleen RT kregen, bedroeg de PFS na twee jaar 30,3%. Toevoeging van DOX deed de PFS na twee jaar stijgen tot 42% (HR = 0,673; P-waarde = 0,002). De verbetering in PFS was consistent in alle vooraf geplande subgroepen. OS na twee jaar in de groep met RT was 41,7%. Door toevoeging van DOX steeg de OS na twee jaar tot 50,8%, een absoluut voordeel van 9% (HR = 0,747; P-waarde was 0,035). Ook hier blijkt het voordeel in OS consistent te zijn in alle vooraf geplande subgroepen. De scores voor QoL waren vergelijkbaar in beide armen.
Deze studie vertoont wel een aantal beperkingen die door dr. Noronha werden aangehaald. In de studie kregen de meerderheid van de patiënten tweedimensionale radiotherapie. 3DCRT of IMRT verlaagt dan wel de incidentie van xerostomie, maar bij patiënten met een niet-nasofaryngeaal carcinoom is niet aangetoond dat dit een grote invloed heeft op de efficaciteit. Daarnaast maakte deze studie geen onderscheid tussen patiënten die adjuvante of definitieve chemoradiotherapie kregen. Ook was dit een studie in één en hetzelfde centrum, al is het dan wel een ziekenhuis met meer dan 10.000 nieuwe hoofd- en halskankerpatiënten per jaar.
Dr. Noronha concludeerde dat de toevoeging van docetaxel aan radiotherapie zowel de ziektevrije overleving als de algehele overleving verbeterde bij patiënten met lokaal gevorderd plaveiselcelcarcinoom van hoofd en hals, die niet in aanmerking komen voor een behandeling met cisplatinum. Volgens haar mag deze behandeling nu gezien worden als een nieuwe referentiestandaard voor zorg bij patiënten die niet in aanmerking komen voor chemoradiotherapie met cisplatinum (3).
Avasopasem manganese: nieuwe behandeling voor orale mucositis?
Dr. Anderson (Universiteit van Iowa) presenteerde de resultaten van de ROMAN-studie, een fase III-studie met avasopasem manganese (AVA) bij patiënten die een ernstige orale mucositis (SOM) ontwikkelen onder chemoradiotherapie (CRT) voor een lokaal gevorderd, niet-gemetastaseerd hoofd- en halscarcinoom (LAHNC).
IMRT + CIS zijn de standaardbehandeling voor LAHNC waarbij een meerderheid van deze patiënten tijdens hun behandeling ernstige orale mucositis zal ervaren. De WHO-scorecriteria voor orale mucositis zijn gebaseerd op zichtbare zweren in de mondholte en op wat de patiënt via de mond kan innemen als dieet. SOM wordt gedefinieerd als graad 3 en 4 orale mucositis. Bij graad 3 noodzaken de zweren in de mond een vloeibaar dieet, bij graad 4 is de patiënt afhankelijk van de voedingssonde. Ongeveer 70% van de patiënten met een LAHNC ontwikkelen een SOM met 20% een graad 4. Mediane duur van de SOM is drie à vier weken, met een mediane start vanaf 40 Gy. Tot op heden is er geen door het FDA goedgekeurd geneesmiddel voor SOM.
Avasopasem manganese is een selectief dismutasemimeticum die een door straling geïnduceerde superoxide snel omzet in waterstofperoxide. Dit is belangrijk omdat superoxide de weefselbeschadiging en ontstekingscascade initieert die resulteert in de ontwikkeling van orale mucositis. In meerdere studies heeft AVA aangetoond dat het normale cellen, maar geen kankercellen, beschermt tegen de schadelijke effecten van straling. AVA toonde veelbelovende resultaten in een gerandomiseerde fase 2-studie, waarbij twee dosisniveaus van 30 mg en 90 mg werden getest versus placebo. De 90 mg dosis verminderde statistisch significant de duur van SOM. Het verminderde ook de incidentie van SOM door bestraling en ook de incidentie van graad 4 OM. Het bijwerkingenprofiel was vergelijkbaar met dat van placebo, en belangrijker nog, de efficaciteit van de behandeling bleef één en twee jaar behouden (4).
De dosis van 90 mg werd gekozen voor de ROMAN-studie, een gerandomiseerde, dubbelblinde fase 3-studie. De studie includeerde patiënten met een lokaal gevorderd plaveiselcelcarcinoom in de mondholte en orofarynx waarvoor IMRT gelijktijdig met CIS was voorzien. De behandeling met AVA 90 mg of placebo bestond uit een IV-infuus van 60 minuten, voorafgaand aan elke stralingsfractie, van maandag tot en met vrijdag, eindigend 60 minuten voorafgaand aan de bestraling. De patiënten werden gestratificeerd op basis van hun chirurgische status en hun cisplatineschema (ofwel een driewekelijks schema ofwel wekelijks 40 mg/m2). Het primaire eindpunt voor deze studie was de incidentie van SOM als gevolg van bestraling. Secundaire eindpunten waren het totale aantal dagen van SOM of de duur, evenals de incidentie en het totale aantal dagen van graad 4 OM.
De studie randomiseerde 241 patiënten in de AVA-groep en 166 in de placebogroep. De patiëntkenmerken waren goed gebalanceerd tussen beide armen, met een meerderheid aan HPV-positieve patiënten, met een orofaryngeale primaire tumor, die definitieve chemoradiatie kregen volgens een wekelijks CIS-schema.
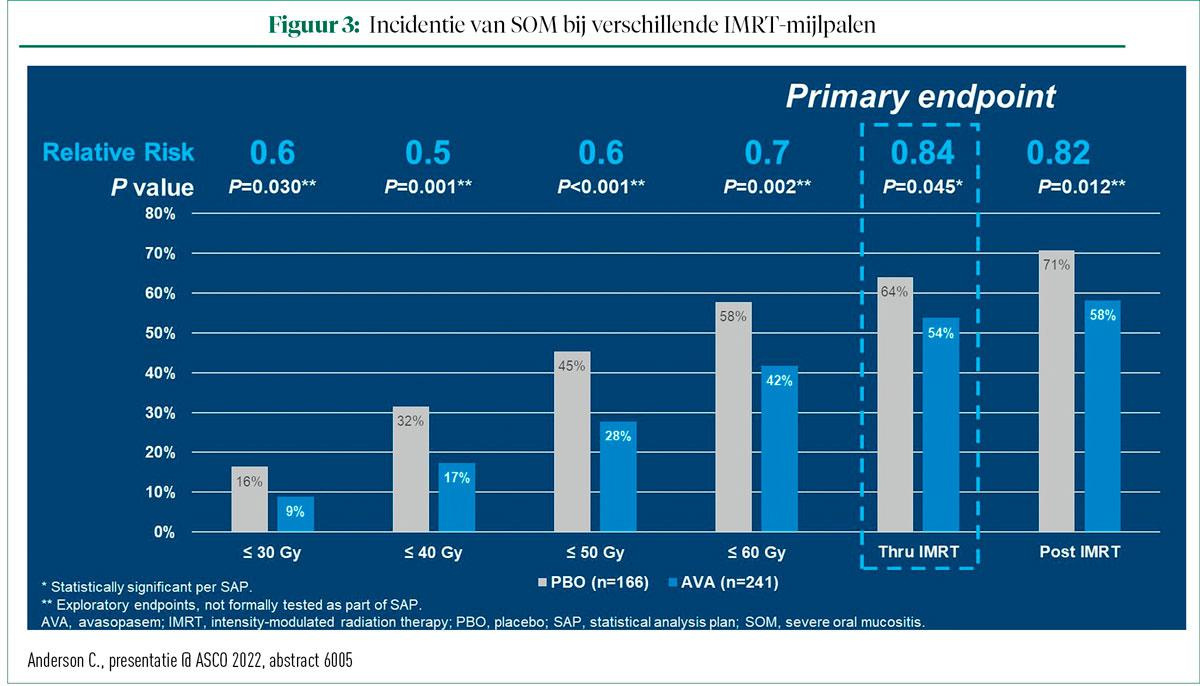
Wat betreft het primaire eindpunt van SOM-incidenten als gevolg van IMRT werd vastgesteld dat AVA de incidentie statistisch verminderde van 64% tot 54% (P-waarde = 0,045). Bovendien verminderde AVA SOM-incidenten zowel gedurende het volledige bestralingsverloop als in de post-IMRT-fase (zie figuur 3).
Er was een indrukwekkende vermindering van 56% in de mediane duur met SOM van 18 dagen in de placebo-arm tot slechts acht dagen in de AVA-arm. De graad 4 OM-incidentie was verminderd, hoewel niet statistisch significant. AVA stelde bovendien de aanvang van SOM uit van 38 dagen in de placebo-arm tot 49 dagen in de AVA-arm. Bij die patiënten die een volledige AVA-kuur kregen, dat wil zeggen 25 infusen placebo of AVA, was de afname in de incidentie van SOM nog aanzienlijker: van 64% met placebo naar 51% met AVA.
AVA 90 mg blijkt goed verdragen te worden, consistent aan de gegevens die de fase II-studie ons leerde. AVA zou wel een milde graad (1 tot 2) misselijkheid en braken veroorzaken, die zelflimiterend is.
Dr. Anderson concludeerde dat AVA 90 mg het eerste geneesmiddel is dat een statistisch significante en klinisch zinvolle vermindering van de incidentie en duur van ernstige orale mucositis laat zien met bovendien ook significante verbeteringen in de ernst en een vertraging in de aanvang. Het veiligheidsprofiel is vergelijkbaar met placebo (5).
1. Hunag et al., Front Oncol, 2020.
2. Jun Ma, presentation at ASCO's Annual Meeting 2022, abstract 6000.
3. Noronha V., presentation at ASCO's Annual Meeting 2022, abstract LBA 6003
4. Anderson et al., JCO 2019.
5. Anderson C., presentation at ASCO's Annual Meeting 2022, abstract 6005.